
PROTACs in Leukemia: A Novel Drug Discovery Strategy from Lab Bench to the Pharmaceutical Market
Andre T. S. Vicente, Laboratory of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Coimbra, Coimbra, Portugal, Center for Neuroscience and Cell Biology (CNC), Center for Innovative Biomedicine and Biotechnology (CIBB), University of Coimbra
Jorge A. R. Salvador, Laboratory of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Coimbra, Coimbra, Portugal, Center for Neuroscience and Cell Biology (CNC), Center for Innovative Biomedicine and Biotechnology (CIBB), University of Coimbra
PROteolysis-TArgeting Chimeras (PROTACs) are a revolutionary targeted protein degradation strategy. To date, several PROTACs that degrade leukemic oncoproteins have successfully overcome many of the limitations of conventional therapies. Although recent, the already enormous potential presented by PROTACs has aroused growing global interest, both from academia and industry, which could translate, sooner or later, into their reaching the pharmaceutical market.
INTRODUCTION
Currently, leukemia remains one of the most common blood malignancies worldwide. In fact, leukemia is a heterogeneous group of life-threatening malignancies that result from the dysfunctional production of abnormal white blood cells in blood-forming tissues, called leukemic cells. These cells exhibit genetic defects, altered functions, and intense proliferative activity, culminating in a series of signs and symptoms characteristic of leukemia.
Leukemia is classified according to their rate of growth, as acute (fast-growing) or chronic (slow-growing), and according to the type of cells from which they originate, known as lymphocytic or myeloid, whether they start from a lymphoblastic cell, or a myeloid cell, respectively. Thus, there are four main types of leukemia: acute lymphocytic leukemia (ALL), acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and chronic lymphocytic leukemia (CLL). There are other types of leukemia, although they are less common.
Nowadays, the most common strategy for treating leukemia is the use of chemotherapy, immunotherapy and/or stem cell transplantation, although the choice always depends on the type of leukemia.
Focusing on chemotherapy, this is carried out using small molecular inhibitors (SMIs). Although it allows obtaining remarkable results, the occurrence of resistance, serious adverse effects, relapses, long-term sequelae, or even constituting a lifelong treatment, lead to a search for new, more effective, and safe therapeutic alternatives.
Among the various innovative therapeutic strategies that have emerged, PROTACs stand out, which are molecules that force the degradation of a protein of interest (POI), thus inactivating its biological function.
Due to their revolutionary event-driven mechanism of action, as well as the advantages that this type of strategy presents compared to the use of inhibitors (Table 1), PROTACs have been the subject of intense research in recent years, not only for the treatment of leukemia, but also in the treatment of other types of cancer, immunological diseases, and neurodegenerative diseases.
PROTACs – What are they?
PROteolysis-TArgeting Chimeras (PROTACs) are bifunctional molecules that allow a selective post-translational degradation of a protein of interest (POI) using the cell’s own machinery; more specifically, the ubiquitin-proteasome system (UPS).
Structurally, PROTACs can easily be divided into three parts, i.e. they present a ligand capable of recruiting the protein of interest (POI) at one end, and a ligand capable of recruiting a UPS element, an E3 ubiquitin ligase, at the other end, these two being connected by a linker.
With this structure, PROTACs simultaneously bind to the target protein and the E3 ligase, forcing them closer together. Because of this forced approach, the E3 ligase catalyses the transfer of ubiquitin moieties to the POI, resulting in its polyubiquitination. By presenting a chain of polyubiquitin on its surface, POI is recognized by the 26S proteasome, which degrades it, thus inactivating its physiological action (Fig. 1).
Historically, the first reported PROTAC is the result of pioneering work by Crews and Deshaies in 2001 that opened the door to targeted protein degradation. Over the past 20 years, the number of PROTACs has increased dramatically, and their evolution can now be divided into three generations. The first generation of large peptide-based PROTACs, followed by a second generation of PROTACs incorporating small chemically synthesized ligands, and the third generation of PROTACs whose action can be controlled in time and space.
More than 20 PROTAC projects are currently in clinical trials worldwide. Highlights include PROTAC ARV-471 from Arvinas and Pfizer, which entered phase III at the end of 2022, for the treatment of breast cancer.
Why are PROTACs so innovative?
Over the past few years, the potential demonstrated by PROTACs has given them tremendous momentum, such that, according to the PROTAC database (PDB), more than 3270 PROTACs have now been reported.
The boom sparked by these degrader molecule therapies is due to several causes. Among them, it is worth highlighting their revolutionary mechanism of action, in which the activity of a POI is suppressed through its forced degradation by PROTAC, and not through its inhibition.
Unlike inhibitors, in which the maintenance of a certain concentration, through high and frequent doses, is required to guarantee inhibition of the target, which often causes adverse effects or resistance, PROTACs only need to establish a temporary connection between the POI and the E3 ligase, to ensure that POI is poly-ubiquitinated and then degraded by the proteasome. After the transfer of ubiquitin units, PROTAC can disconnect from the POI, and is free to bind to the new target protein, thus guaranteeing it a catalytic mechanism of action (event-driven mechanism), and in turn reducing the drug's toxicity and improving its side effect profile.
Another great advantage of PROTACs is that these molecules can bind to any domain of the target since they only have to guarantee the proximity of the POI to the E3 ligase. This means that proteins that do not have an active site (so-called “undruggable proteins”, such as transcription factors or scaffold proteins) can now be targeted, opening a whole Pandora’s box of possible new therapeutic targets for already known diseases.
Furthermore, these molecules can eliminate intracellular protein aggregates, do not cause compensatory expression of POI, and are less susceptible to the development of resistance. PROTACs also have a series of other advantages to be discussed later in this article.
PROTACs ̶ the race for discovery
With the consolidation of the potential of PROTACs, the race to discover better degraders has been intensifying, resulting in an exponential increase in the number of scientific publications, patent applications, research groups dedicated to the study of PROTACs and even in founding of several companies dedicated to its commercialization.
Arvinas, co-founded by Dr. Crews, is one of the first companies to begin commercial development of PROTACs. Currently, it has a strong portfolio of PROTACs, with several PROTACs already in different phases of clinical trials, with emphasis on PROTAC ARV-471 (phase III).
Over the years, other companies dedicated to the commercialization of PROTACs were also founded. For example, in 2012, Nurix therapeutics emerged, which already has three PROTACs in phase I. Other examples of companies with PROTACs in clinical trials are Kymera therapeutics, C4 Therapeutics, Bristol Myers Squibb, Haisco, Accutar Biotech, among others. Some of the previous ones have partnerships with big pharmaceutical companies, such as Pfizer, Sanofi, or Merck, which demonstrates the large investment that this type of targeted protein degradation has raised, a sign of its potential.
Anti-leukemic PROTACs
PROTACs have been studied in a wide variety of diseases. However, the focus has been on the treatment of different types of cancer, including leukemia.
Nowadays, there are already several reported PROTACs capable of promoting the degradation of targets involved in the development and progression of distinct types of leukemia. The oncoproteins BCL-XL, BCR-ABL, BRD4, BTK, CDK6, FLT-3, among others, are considered the main targets to be degraded by anti-leukemia PROTACs (Fig. 2).
Currently, the overwhelming majority of anti-leukemic PROTACs are in preclinical development. However, it is already possible to highlight some promising degraders that fulfil the purpose of potently degrading the target, and thus have a potential in the future treatment of one or more types of leukemia.
Degradational studies demonstrated that some of the anti-leukemic PROTACs have been shown to achieve potent, rapid, and time-sustained degradation of the intended targets in vitro and in vivo. Most notably, in 2017, Wang's laboratory reported the compound BETd-260 capable of degrading BRD4 with picomolar potency in leukemia cells, in a time- and concentration-dependent manner. Most of the reported lead PROTACs present half-maximal target degradation (DC50) values in the nano-molar range, such as the BCR-ABL PROTAC GMB-805 (340 nM), the BCL-XL PROTAC DT2216 (63 nM), BRD4 PROTAC ARV-825 (5 nM). Furthermore, in some cases, such as BCR-ABL PROTAC SIAIS178, the anti-leukemic action lasts longer after its removal than the isolated inhibitor. It is also important to note that the maximum level of degradation of these PROTACs is around 80 to 100%.
A remarkable advantage presented by some of the reported anti-leukemic PROTACs is a greater selectivity for the target and cell type, which translates into a potential reduction in adverse effects. The BCL-XL PROTAC XZ739 showed 100 times greater selectivity for MOLT-4 leukemia cells over platelets, while the navitoclax inhibitor presented cytotoxicity against both cell types, reducing the risk of the degrader causing thrombocytopenia. Another example of an improved selectivity of PROTAC when compared to the isolated inhibitor was the study presented by Rao's group, where all palbociclib-based PROTACs are much more selective for CDK6 than the respective isolated palbociclib inhibitor. This improved selectivity of PROTACs is explained by the new protein-protein interactions that are established during the formation of the ternary complex (target-PROTAC-E3 ligase), favouring the binding of PROTAC to a given target, to the detriment of another.
One of the major difficulties associated with the use of inhibitors is the frequent and unpredictable occurrence of mutations, which lead to the triggering of resistance, making it impossible to use the inhibitor for the treatment of a certain type of leukemia. However, there are several anti-leukemic PROTACs that have been shown to selectively degrade oncoproteins with resistance-conferring mutations. In 2018, a FLT3 PROTAC was reported capable of degrading, in vivo, one FLT3 mutant form with nanomolar potency, causing a 60% decrease in FLT-3 levels compared to the control level. In 2019, Zhao et al. presented the BCR-ABL PROTAC SIAIS178, that when tested on several clinically relevant mutant isoforms that confer resistance to imatinib or dasatinib, was shown to degrade some of the mutant forms of BCR-ABL, on the other hand, the inhibitor demonstrated very reduced activity. In 2022, another BCR-ABL PROTAC, SIAIS100, caused a significant reduction in the levels of the T315I and E255V mutant forms, as well as some asciminib-resistance mutations, in different leukemic cells. This great advantage comes from the fact that PROTACs only have to bind to the target protein, regardless of the binding site, and bring it closer to the E3 ligase.
From the degradation of these leukemic oncoproteins by PROTACs, it is possible to observe, in a first phase, the triggering of inactivation of the target's action and a blockage of its signalling pathway, and in a second phase, a reduction in cell proliferation, and activation of cell apoptosis, culminating in a powerful anti-leukemic effect. For example, in vivo studies demonstrated that BRD4 PROTAC dBET1 has a superior antileukemic effect than JQ1 inhibitor. Administration of dBET1 reduced tumour progression in murine xenograft model of human AML cells, accompanied by degradation of BRD4 and downregulation of c-Myc. Another example is PROTAC DT2216, that in vivo studies demonstrated that it is not only safer, but also more potent than navitoclax inhibitor. Furthermore, when tested in vivo, the BET degrader ARV-771 was more potent in reducing leukemic burden and increasing survival rate than the inhibitor OTX015.
In addition to the targets discussed previously, new potential PROTACs have recently emerged with a view to inducing chemical knockdown of new therapeutic targets important in the development of different types of leukemia, for example, MDM2, STAT3/5, HDACs or SMARCA2/4.
According to the Clinicaltrials.gov database, there are currently six anti-leukemic PROTACs in phase I of clinical trials, most for the treatment of B-cell malignancies (Table 2), including PROTAC DT2216. It should be noted that most PROTACs under clinical studies aim at the forced degradation of the BTK proteins.
PROTACs in leukemia – Future Perspectives
Some of the anti-leukemic PROTACs presented to date have shown excellent selectivity against targets relevant to the development of leukemia, enabling a leap away from the current inhibitor-based drug paradigm.
Enormous progress has been made in the development of anti-leukemic PROTACs, and there are already several PROTACs with potential for the treatment of one or more types of leukemia, some of which are already in clinical trials and have demonstrated that they can overcome some of the main difficulties associated with the use of inhibitors, such as the emergence of resistance. However, it should be noted that there is still a long way to go, with many aspects to be studied and clarified, as well as some challenges to overcome, such as the large size of this type of molecules.
In summary, we can say that we are facing with very promising molecules with enormous potential for scientific innovation that could one day reach the pharmaceutical market and add value to the future treatment of one or more types of leukemia.
Table 1. Comparisons of PROTAC with small molecule inhibitors (SMIs).
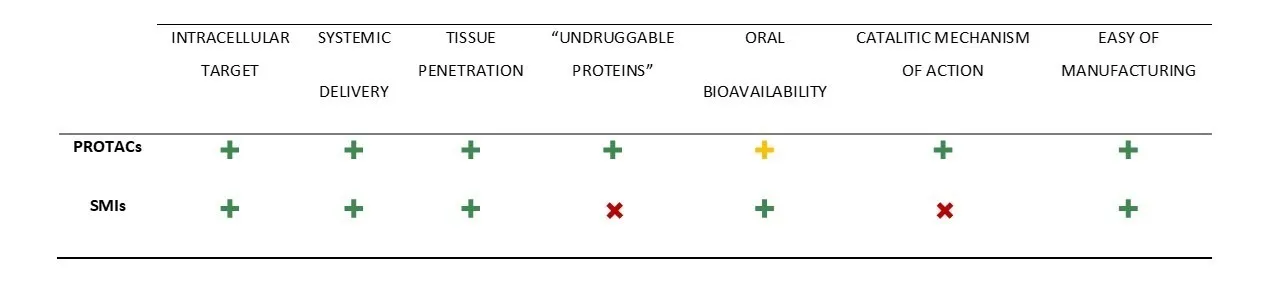
Table 2. PROTACs for the leukemia treatment in Clinical Trials - from the Clinicaltrials.gov database.
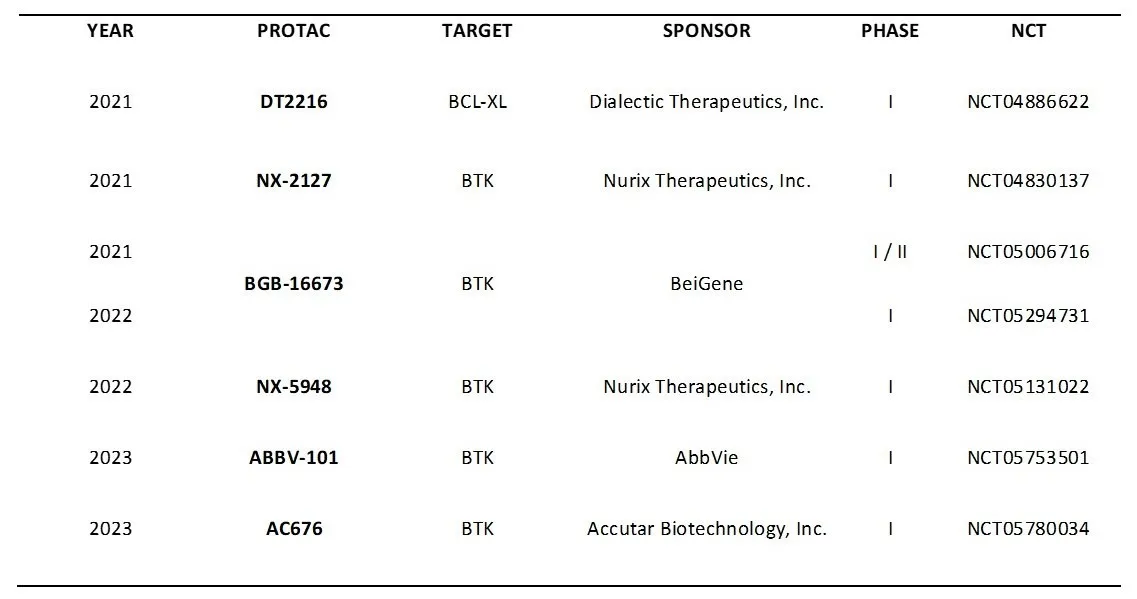
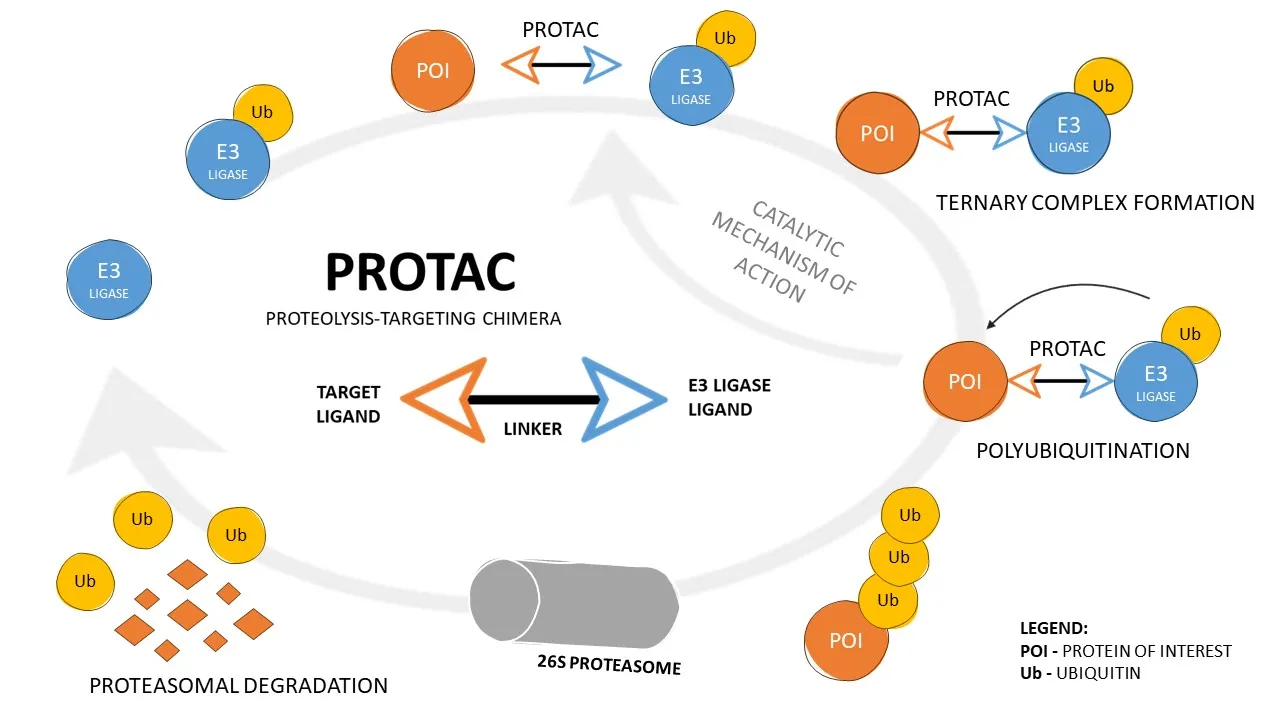
Figure 1. Mode of action of PROteolysis-TArgeting Chimeras (PROTACs).
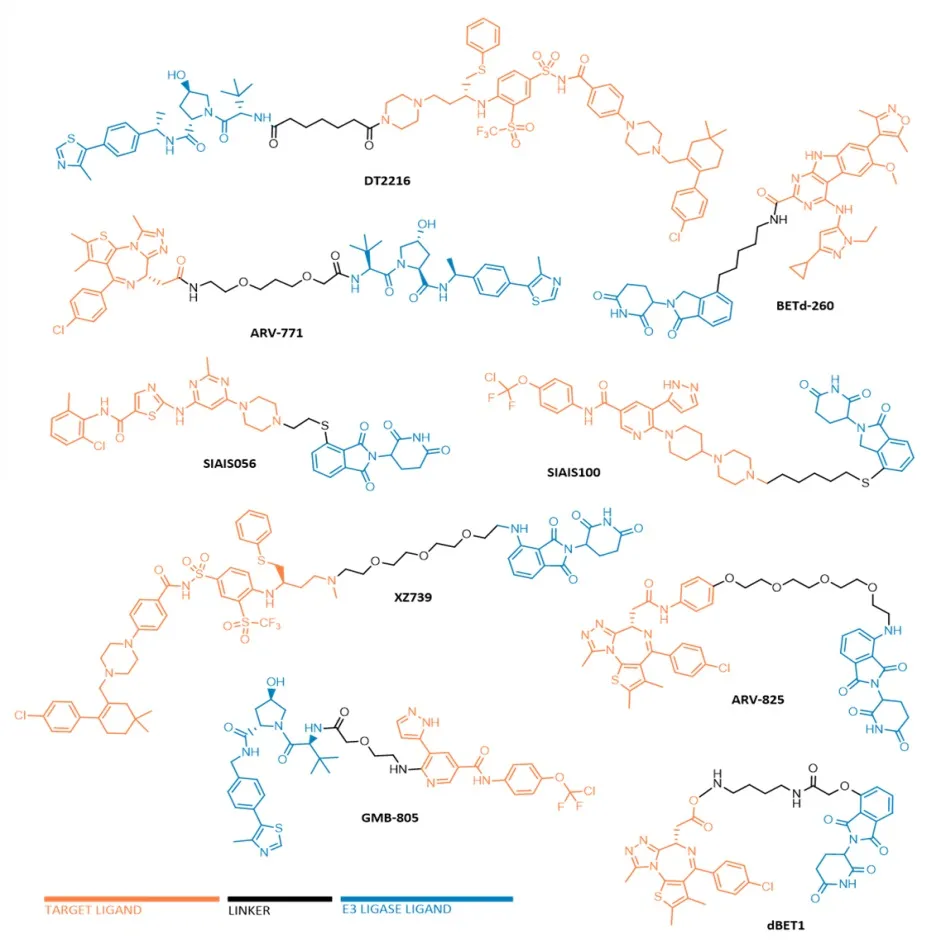
FIGURE 2. Chemical structures of some anti-leukemic PROTACs.
REFERENCES
- Vicente, A.T.S., and Salvador, J.A.R. (2022) MDM2-Based Proteolysis-Targeting Chimeras (PROTACs): An Innovative Drug Strategy for Cancer Treatment. Int. J. Mol. Sci., 23 (19), 11068.
- Vicente, A.T.S., and Salvador, J.A.R. (2023) Proteolysis-Targeting Chimeras (PROTACs) targeting the BCR-ABL for the treatment of chronic myeloid leukemia – a patent review. Expert Opin. Ther. Pat., 33 (6), 397–420.
- Bou Malhab, L.J., Alsafar, H., Ibrahim, S., and Rahmani, M. (2023) PROTACs: Walking through hematological malignancies. Front. Pharmacol., 14, 1–10.
- He, Y., Khan, S., Huo, Z., Lv, D., Zhang, X., Liu, X., Yuan, Y., Hromas, R., Xu, M., Zheng, G., and Zhou, D. (2020) Proteolysis targeting chimeras (PROTACs) are emerging therapeutics for hematologic malignancies. J. Hematol. Oncol., 13 (1), 103.
- Anwar, Z., Ali, M.S., Galvano, A., Perez, A., La Mantia, M., Bukhari, I., and Swiatczak, B. (2022) PROTACs: The Future of Leukemia Therapeutics. Front. Cell Dev. Biol., 10.
- Békés, M., Langley, D.R., and Crews, C.M. (2022) PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov., 21 (3), 181–200.
- Zhou, B., Hu, J., Xu, F., Chen, Z., Bai, L., Fernandez-Salas, E., Lin, M., Liu, L., Yang, C.Y., Zhao, Y., McEachern, D., Przybranowski, S., Wen, B., Sun, D., and Wang, S. (2018) Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem., 61 (2), 462–481.
- Saenz, D.T., Fiskus, W., Qian, Y., Manshouri, T., Rajapakshe, K., Raina, K., Coleman, K.G., Crew, A.P., Shen, A., Mill, C.P., Sun, B., Qiu, P., Kadia, T.M., Pemmaraju, N., Dinardo, C., Kim, M.S., Nowak, A.J., Coarfa, C., Crews, C.M., Verstovsek, S., and Bhalla, K.N. (2017) Novel BET protein proteolysis-Targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia, 31 (9), 1951–1961.
- Lu, J., Qian, Y., Altieri, M., Dong, H., Wang, J., Raina, K., Hines, J., Winkler, J.D., Crew, A.P., Coleman, K., and Crews, C.M. (2015) Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol., 22 (6), 755–763.
- Winter, G.E., Buckley, D.L., Paulk, J., Roberts, J.M., Souza, A., Dhe-Paganon, S., and Bradner, J.E. (2015) Phthalimide conjugation as a strategy for in vivo target protein degradation. Science, 348 (6241), 1376–1381.
- Zhang, X., Thummuri, D., Liu, X., Hu, W., Zhang, P., Khan, S., Yuan, Y., Zhou, D., and Zheng, G. (2020) Discovery of PROTAC BCL-XL degraders as potent anticancer agents with low on-target platelet toxicity. Eur. J. Med. Chem., 192, 112186.
- Khan, S., Zhang, X., Lv, D., Zhang, Q., He, Y., Zhang, P., Liu, X., Thummuri, D., Yuan, Y., Wiegand, J.S., Pei, J., Zhang, W., Sharma, A., McCurdy, C.R., Kuruvilla, V.M., Baran, N., Ferrando, A.A., Kim, Y., Rogojina, A., Houghton, P.J., Huang, G., Hromas, R., Konopleva, M., Zheng, G., and Zhou, D. (2019) A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med., 25 (12), 1938–1947.
- Liu, H., Mi, Q., Ding, X., Lin, C., Liu, L., Ren, C., Shen, S., Shao, Y., Chen, J., Zhou, Y., Ji, L., Zhang, H., Bai, F., Yang, X., Yin, Q., and Jiang, B. (2022) Discovery and characterization of novel potent BCR-ABL degraders by conjugating allosteric inhibitor. Eur. J. Med. Chem., 244 (October), 114810.
- Burslem, G.M., Bondeson, D.P., and Crews, C.M. (2020) Scaffold hopping enables direct access to more potent PROTACs with in vivo activity. Chem. Commun., 56 (50), 6890–6892.
- Zhao, Q., Ren, C., Liu, L., Chen, J., Shao, Y., Sun, N., Sun, R., Kong, Y., Ding, X., Zhang, X., Xu, Y., Yang, B., Yin, Q., Yang, X., and Jiang, B. (2019) Discovery of SIAIS178 as an Effective BCR-ABL Degrader by Recruiting Von Hippel–Lindau (VHL) E3 Ubiquitin Ligase. J. Med. Chem., 62 (20), 9281–9298.

André Vicente has a master’s degree in Pharmaceutical Sciences from Faculty of Pharmacy of the University of Coimbra (FFUC). Currently, he is enrolled in the doctoral program in Pharmaceutical Sciences at FFUC. His research work has focused on the design and study of PROteolysis-TArgeting Chimeras (PROTACs) to promote the targeted degradation of oncogenic proteins

Jorge Salvador is Full Professor at the Faculty of Pharmacy of the University of Coimbra and is the group leader of the research group “Medicinal Chemistry & Drug Discovery” at the Centre for Innovative Biomedicine and Biotechnology. His work has been focused on studies of new anticancer compounds, including the study of PROTACs.





