

How to Study Drug Transport at Biological Interfaces
Laura Housman, Practice Director at Avalere
Mohammad Sharifian, Department of Cell Biology, University of Virginia
To design drugs with better absorption and distribution, scientists need to have a comprehensive understanding of the transport mechanism of drugs at biological interfaces. Thus, developing techniques capable of monitoring transport in real-time is crucial for pharmaceutical industries. Currently, fluorescence, SPR, and SHS are the most state-of-the-art approaches.
Drugs as small- or medium-sized molecules (1-2 nanometer in diameter) typically possess both lipophilic and hydrophilic properties. For a drug molecule to reach its intracellular target, it usually needs to cross several barriers including mucus gel layer, intestinal epithelial cells, capillary endothelium, and finally membrane of target cells. In all those interfaces, lipoidal diffusion and protein-mediated passive transport across lipid bilayers play an important role in its molecular entry. Thus, understanding the mechanism of drug transport at biological membranes provides us with a theoretical foundation for designing powerful drugs with better Absorption, Distribution, Metabolism, Excretion, and Toxicology (ADMET) properties.
Physicochemical properties of drug molecules, including molecular weight, surface charge, lipophilicity, acid/base properties, number of hydrogen bonds and rings determine their behavior in a solution and at membrane barriers. While the ‘Overton’s rule’ simply states that membrane permeability of a permeant, Pm (m s-1) is proportional to the product of its diffusivity, Dp (m2 s-1) and oil/water partition coefficient, Ko/w (unitless), in a quantitative structure-permeability relationship (QSPR) model, drug’s molecular weight, number of aromatic or non-aromatic rings, and hydrogen bond donors and acceptors are utilised to predict permeability of the molecule by which poor permeability is more likely for Ko/w> 100,000, molecular weight > 500 Da, hydrogen bond donors > 5, and hydrogen bond acceptor > 10 1.
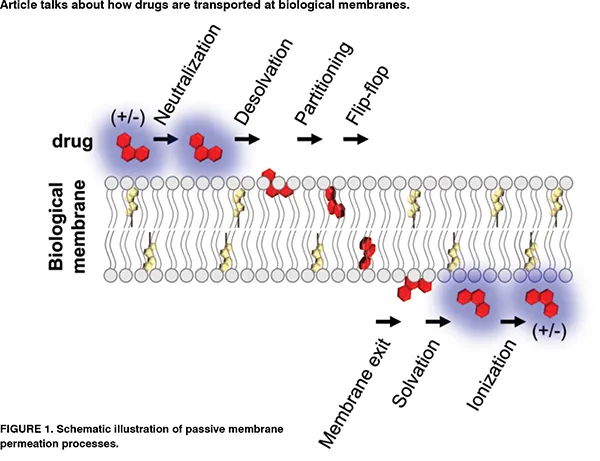
The anisotropic and inhomogeneous nature of biological membranes has encouraged scientists to develop ‘inhomogeneous solubility-diffusion model’ which considers depth-dependent partitioning, resistance, and diffusion parameters. As depicted in Figure 1, drug permeation is determined by two main equilibria of drug partitioning between the aqueous compartments and the lipid leaflets, and drug translocation between the two lipid leaflets by which for lipophilic compounds, the membrane may act as both a barrier and a sink. The following equation can be used to estimate the permeability of drug molecules at a lipid bilayer of biological membranes2, where, is the membrane viscosity (kg m-1 s-1), dm is the effective thickness of the membrane, Vp is the volume of the drug molecule (m3), is the size selectivity factor of the permeant, Kc ⁄w is the partition coefficient of drug between water and chloroform, and kB and T are respectively Boltzmann constant (1.38×10-23 m2 kg s-2 K-1), and temperature (K).
Model Membrane Systems in Drug Permeation Measurements
A monolayer of cultured colorectal adenocarcinoma-derived cells (Caco-2 cell permeability assay) is widely used to mimic a single layer of intestinal epithelium in drug permeation measurements. In this approach, cells are grown on a porous filter separating two stacked microwells with the drug candidate added to one of chambers, and its transport through the interface is then probed, usually by fluorescencebased techniques. Due to complexity of the Caco-2 cellular membranes, and the time required for the preparation of stable monolayers of the cells in each experiment (up to 30 days), scientists prefer to use model membranes which also allow them to perform experiments under conditions that Caco-2 cells may not withstand. Moreover, they would be able to apply a variety of methods for permeation measurements, perform high-throughput analyses, and discern the roles of lipids in drug permeation processes. Model membranes include: 1.) Parallel artificial membrane permeability assay (PAMPA), 2.) Phospholipid vesiclebased permeation assay (PVPA), and 3.) liposomes in suspension. Similar to Caco-2 cell assay, in PAMPA and PVPA, porous filter membranes are used to separate two stacked microwells. Liposomes, including small (SUV; 20-100 nm), large (LUV; 0.1-1 m), and giant (GUV; 1-100 m) are biomimetic model systems that can reliably reproduce the bilayer structure. They also allow scientists to apply various techniques in their studies. Note that model membranes lack typical cell membrane characteristics such as membrane proteins, and therefore, biophysical interactions of drug molecules with model membranes may not exactly replicate all the aspects of the cellular uptake processes. Thus, for an accurate extrapolation of in vivo drug permeation measurements, researchers should select a suitable membrane for the purpose in mind.
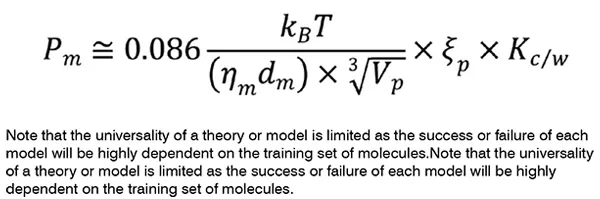
Novel Approaches in Drug Permeation Measurements
Fluorescence-based techniques, surface plasmon resonance (SPR), and secondharmonic light scattering (SHS) are the most recent approaches to study drug transport at biological interfaces 2.
Both spectroscopic and imaging modalities of fluorescence techniques have been applied in a wide range of model systems (PAMPA, Caco-2 cells, or liposomes) by which either the drug molecule is fluorescing under some conditions (e.g., at a specific pH) or a fluorescent probe is used to track the transport of the drug molecule (indirect measurement) (see Figure 2A). In microscopic modality, transport of the drug molecule can even be visualised with sub-liposomal spatial resolution. Developing a phenomenological model in which membrane permeability of the drug is related to its concentration in the region of measurement (i.e., signal intensity is proportional to the concentration of the drug) will allow us to deduce membrane permeability value along with kinetic parameters, e.g., rate constants of sequential processes. Fluorescence techniques have been widely used to monitor the transport of anti-cancer drugs and tetracycline antibiotics across liposomal membranes, and to determine the antibiotic uptake in bacteria at individual cell level. Fluorescence techniques can also be combined with other approaches such as reflectometric interference spectroscopy.
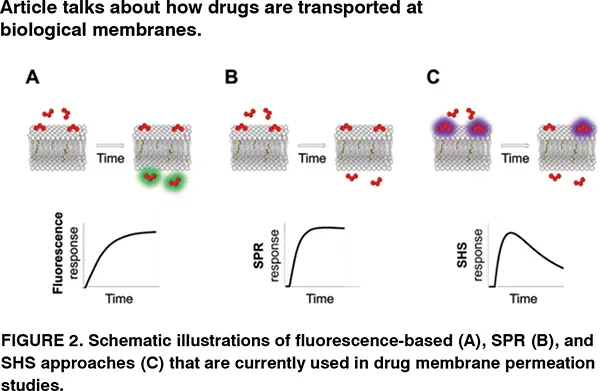
In contrast to fluorescence, SPR is a label-free technique that is based on evanescent wave sensing by which a polarised light beam reflected from the sensor chip (gold or silver) is absorbed by the free electrons at the surface causing them to resonate (surface plasmon). Any reaction occurring close to a metal surface leads to altered conditions for surface plasmon excitation. Interaction of drug molecules with the immobilised liposomes on the sensor surface can be followed in real-time by measuring an optical response, usually expressed in response units, RU (see Figure 2B). A phenomenological model can relate the membrane permeability of the drug molecule with the time-resolved SPR responses, for example, to screen melittin- and aquaglyceroporin PfAQPmediated transfer of sugar alcohols and sucrose across liposome membranes. SPR has the advantage of being easily combined with micro-fluidic devices, and perform multiple measurements on the same set of immobilised liposomes.
SHS is a label-free, real-time, and membrane-specific optical technique with high sensitivity. It, in contrast to fluorescence, involves coherent radiative scattering where incident light of frequency induces a polarisation of frequency 2 serving as a source of the SHS scattered light. In a typical experiment, a sample of an SH-active drug molecule is exposed to an incident laser beam supplied by a mode-locked Ti:Sapphire laser, and the scattered signal is selectively collected by using a band-pass filter and monochromator. Note that the SHS light from drug molecules in solution destructively adds to zero, but when molecules adsorb onto the membrane, they align with one another resulting in a coherent signal that scales quadratically with the drug concentration at the membrane surface. If the molecule diffuses across the membrane, signals from the oppositely oriented molecules on the inner surface cancel with the signal from the outer surface, which results in a real-time decrease of the signal (see Figure 2C). By subjecting the time-resolved SH signals to a phenomenological model, rate value of each step can be quantified. SHS has been applied to monitor the diffusion of druglike molecules across lipid membranes in liposomes and living cells, to study druginduced changes to membrane properties, and to quantify molecular transport in sub-cellular regions of individual living cells.
Overall, in modern drug discovery research, recently developed methodologies including fluorescence, SPR, and SHS can be exploited to elucidating how the molecular structure of the drug candidates relates to their membrane permeability.
Author Bio

Laura Housman is a Practice Director at Avalere and leads the Evidence & Strategy practice, applying her background in evidence generation, commercialization, and strategy to a broad range of projects. Before joining Avalere, Housman held a variety of leadership positions in industry and academia, including at Novartis, Exact Sciences, and BlueCross BlueShield of Massachusetts.

Working as a Postdoctoral Research Associate in the Laurie lab at University of Virginia (Cell Biology) immediately after receiving Ph.D. in Physical Chemistry (Temple University), Mohammad is currently studying the antimicrobial mechanism of tear lacritin peptide ‘N-104’ towards ocular pathogens






