
Current state
Antibody drug conjugates (ADCs) are innovative modalities to deliver chemotherapeutics to specifically targeted populations of cancer cells. The first ADC approved by the United States Food and Drug Administration (FDA) in 2000 was gemtuzumab ozogamicin developed by Celltech and Wyeth for treatment of patients with Acute Myeloid Leukemia (AML). Currently, there are 12 FDA-approved ADCs on the US market for treatment of hematologic and solid tumors (Table 1). The basic purpose of this novel modality is to increase efficacy of anti-neoplastic therapies and to reduce their toxic side effects. Toxicity of chemotherapy often prevents dosing drug to an effective level or leads to a reduction in drug dose or cessation of a treatment. ADCs aim to deliver chemotherapeutics at an effective level combined with elimination or reduction of dose-limiting toxicity.
Typically, ADCs comprise a monoclonal antibody (mAb), a payload molecule e.g., “a small molecule” chemotherapeutic and a linker between the two (Figure 1). The mAb guides the drug to the population of tumor cells expressing a unique, targeted epitope absent on the surface of healthy cells. This combination of specificity of the mAb and the tumor marker provides a mechanism for reduction of off-target toxic effects of the chemotherapy and allows an increase of the drug efficacy. The linker stably attaches the payload to the mAb without affecting the mAb's ability to bind to its target and allows the payload to be released from the ADC upon reaching the intended cancerous cell. Mechanisms by which payload molecules can kill tumor cells include DNA cleavage and cross-linking (calicheamicin, pyrrolobenzodiazepine dimer), microtubule inhibition (auristatin, maleimidocaproyl monomethyl F, maytansine, maytansinoid DM4), topoisomerase inhibition (Deruxtecan, SN-38) and induction of apoptosis (Pseudomonas exotoxin A). Table 1 lists malignancies targeted by ADCs currently in the clinic. Patient populations presenting with advanced or metastatic tumors that were not responsive to one or more of the conventional chemotherapies often receive ADC therapies.
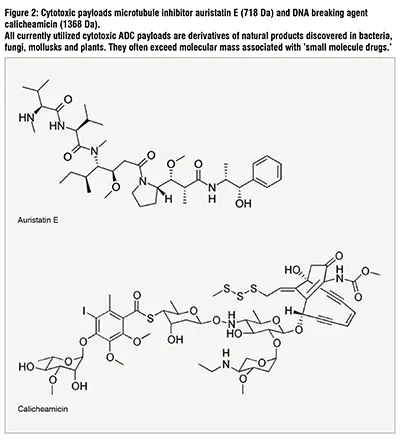
The potential toxicity of the ADC therapies is a recognized concern. Nine out of 12 ADCs on the market carry a boxed warning. Boxed warnings, formerly known as Black Box Warnings, are the highest safety warnings that the FDA assigns to medications. The toxicities observed in clinical trials can vary among ADCs carrying the same payload. Four ADCs utilize auristatin E as a chemotherapeutic agent: brentuximab vedotin, tisotumab vedotin, polatuzumab vedotin and enfortumab vedotin (Figure 2, Table 1). Only the former two required boxed warnings; brentuximab vedotin for progressive multifocal leukoencephalopathy in Hodgkin’s lymphoma, systemic anaplastic large cell lymphoma and HER2-positive, metastatic breast cancer patients; and tisotumab vedotin for changes in the corneal epithelium and conjunctiva resulting in changes in vision, including severe vision loss, and corneal ulceration in patients with recurrent or metastatic cervical cancer.
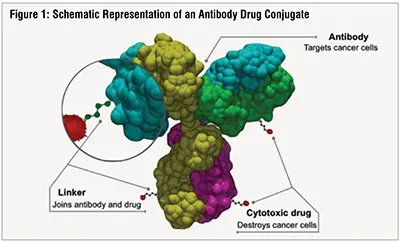
(Source of the image - https://commons.wikimedia.org/wiki/File:Antibody-drug_conjugate_structure.svg)
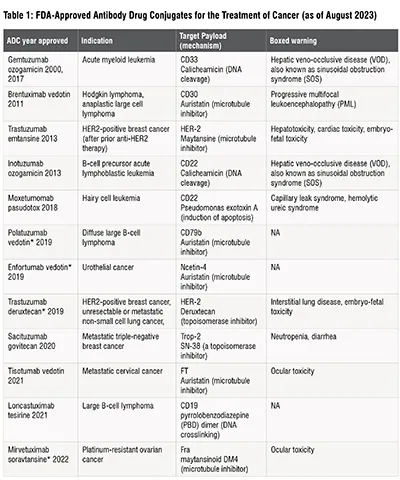
*- accelerated approval
NA – not applicable
Not listed above is belantamab mafodotin approved in 2020 for treatment of adult patients with relapsed or refractory multiple
myeloma but withdrawn in 2022 due to the lack of efficacy.
The diversity of toxicities between brentuximab vedotin and tisotumab vedotin suggests that the individual components of an ADC as well as disease and genetic characteristics of the patient population are jointly responsible for the findings. Intrinsic factors such as renal or liver impairment, pharmacogenomics, body weight, age, gender, and race have potential to influence clinical pharmacology of an ADC (Clinical Pharmacology Considerations for Antibody-Drug Conjugates, Guidance for Industry, FDA, 2022, draft). ADC immunogenicity and drug-drug interactions (DDI) can have similar potential to influence the exposure of the medication.
In addition to the two ADCs containing auristatin E, gemtuzumab ozogamicin and inotuzumab ozogamicin that utilize calicheamicin as their payload, also carry boxed warning for the same toxicity, namely hepatotoxicity (Figure 2, Table 1). Gemtuzumab ozogamicin is approved for treatment of newly diagnosed CD33-positive AML and for treatment of relapsed or refractory CD33-positive AML, and inotuzumab ozogamicin is approved for treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL), caused hepatotoxicity. The toxicity includes severe or fatal hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome (SOS). These severe adverse reactions result in dose reduction and can lead to discontinuation of treatment and therefore deprive the patients of the modality’s promise of delivering a precisely targeted and effective dose of chemotherapeutic agent.
ADCs are large molecules, typically >150,000 molecular weight mass, which may present multiple safety issues. Safety considerations presented by ADCs include DDI potential of the payload molecule. Specifically, the ADC may act as a DDI precipitant through the effects of the payload molecule on the mRNA expression or enzymatic activity of drug metabolizing enzymes and on drug transporters. These effects of payload molecules are a major concern to be addressed during the drug development process and are the focus of our discussion. The mAb portion of the ADC is proteolytically degraded to small peptides and individual amino acids in lysosomes, and therefore generally it’s not considered to be a risk factor for the DDI. It’s noteworthy that the trastuzumab deruxtecan antibody portion itself may contribute to interstitial lung disease observed in patients treated with the ADC (SOT 2023 Annual Meeting, abstract 5057). A fragment of the linker that finds its way to plasma may have the potential to contribute to DDI. An unwanted degradation of an ADC in plasma rather than inside of the target cell could lead to systemic exposure to the toxic payload. Metabolic stability of the ADC payload in plasma, in the lysosomes and the cytosol is of concern from the perspective of toxicity and efficacy. The payload, typically an extremely potent toxin, needs to retain its potency while transitioning through the lysosomal compartment and should not be freely present in the plasma, as it may enter healthy cells. But the payloads selected for stability in these environments present an additional challenge as they may still be active when the content of the killed cancer cells is released. Other safety considerations for the payload molecules include long half-life, bystander killing activity, systemic accumulation, and potential for development of tumor resistance.
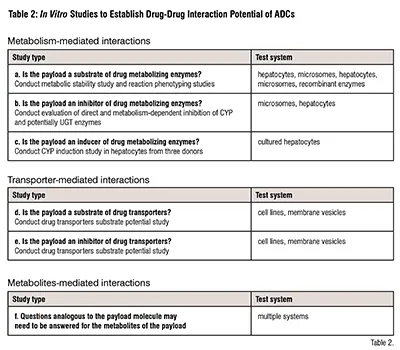
Our review of the package inserts found no indication of ADC being a perpetrator of DDI in vivo. In vitro studies addressing DDI perpetrator potential of the ADC are presented below (Table 2). These studies were conducted on a case-by-case basis, as they may not be necessary for repeated use of payloads or the payload chemistry, e.g., Pseudomonas exotoxin A, a peptide, is not expected to be a CYP enzyme inhibitor or an inducer. An ADC can be a victim of DDI, also referred to as an object of DDI, when metabolism of its payload is changed by co-administered drugs, e.g., inhibitors or inducers of drug metabolizing enzymes. In vitro metabolism of the payloads, followed by reaction phenotyping studies, elucidate victim potential of ADCs (Table 2).
Drug-drug interactions involving ADC
Auristatin E. Various ADC currently in pre-clinical and clinical development and in the clinic (brentuximab vedotin, polatuzumab vedotin, enfortumab vedotin and tisotumab vedotin), are designed to treat several forms of cancer utilizing auristatin E as their payload. In vitro data indicate that monomethyl auristatin E (monomethyl auristatin E, MMAE) is a substrate and an inhibitor of CYP3A4/5. In vitro data indicate that MMAE is also a substrate of the efflux transporter P-glycoprotein (P-gp). These in vitro data indicated a perpetrator and a victim potential for the auristatin E-containing ADC. A victim potential was considered due to the molecule being extensively metabolized by CYP3A4. The in vitro studies were followed up with clinical studies. For brentuximab vedotin it was demonstrated that the ADC did not affect exposures of sensitive CYP3A4 substrate midazolam. ADC exposures were unaffected by concomitant rifampin or ketoconazole; however, the auristatin E exposures were lower with rifampin and higher with ketoconazole consistent with the molecule being metabolized by CYP3A4. The study demonstrated the low magnitude of the interaction such that dose adjustments were not required (J Clin Pharmacol. 2013 August; 53(8): 866–877). Similar interactions can be expected for other ADC delivering auristatin E.
Sacituzumab govitecan. An SN-38, an active metabolite of anticancer drug irinotecan, is a payload of Sacituzumab govitecan. The SN-38 is inactivated by UDP-glucuronosyl transferase UGT1A1, therefore the ADC can be a DDI victim when co-administered with UGT1A1 inhibitors which would lead to a toxic level of the payload molecule. Concomitant administration of sacituzumab govitecan with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38. Exposure to SN-38 may be reduced in patients concomitantly receiving UGT1A1 enzyme inducers. The package insert recommends avoiding administration of sacituzumab govitecan with UGT1A1 inhibitors or inducers (sacituzumab govitecan package insert). Use of sacituzumab govitecan presents an analogous risk of toxicity in patients who are genetically deficient in the activity of UGT1A1, e.g., homozygote UGT1A1*28. Interactions of sacituzumab govitecan with UGT inducers have not been identified, presumably due to a relatively low level of inducibility of this family of enzymes as compared to cytochrome P450 enzymes (CYP).
Inotuzumab ozogamicin. A potential for non-pharmacokinetic DDI has been identified in the ADC label. Concomitant use of inotuzumab ozogamicin with drugs known to prolong the QT interval or induce Torsade de Pointes may increase the risk of a clinically significant QTc interval prolongation. Discontinuation or use of alternative concomitant drugs that do not prolong QT/QTc interval while the patient is using inotuzumab ozogamicin was recommended (inotuzumab ozogamicin package insert).
In vitro DDI evaluation considerations
To assure safety of the ADC, regulatory agencies recommend certain in vitro studies of their potential DDI. The studies can be based on the FDA Guidance “In vitro Drug Interaction studies – Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions” (2020) and the draft “Drug Interaction Studies M12” guidance issued in 2022 by the International Council for Harmonization (ICH). The ICH guidance is expected to be finalized in 2024. The two regulatory documents focus on small molecule drugs and therefore apply to evaluating payloads of the ADC. Two important points presented in the draft ICH M12 guidance regarding potential DDI of the ADCs are –
- For ADCs, the small molecule drug component conjugated to the antibody component can be released in unconjugated form. Therefore, the DDI potential of both the antibody and the small molecule drug component should be considered. In general, for the small molecule component, the potential to inhibit or induce enzymes and transporters should be addressed in line with what is described elsewhere in this guideline. In many cases, however, the systemic concentration of free drug might be too low to act as a perpetrator in vivo.
- It is important to understand the formation, distribution, and elimination kinetics of the small molecule and to assess the systemic exposure of the small molecule drug component of the ADC. It might be necessary to evaluate the small molecule component (administered as an ADC) as a victim drug, in particular if increased levels of free drug may be associated with safety concerns. Understanding the exposure-response relationship of the various moieties is important in determining whether to conduct DDI studies and their significance.
Mechanisms of DDI include metabolism- and transporter-dependent processes that may involve the parent compound and its metabolites. Table 2 presents types of studies and their respective appropriate test system used for addressing questions of DDI potential of chemotherapeutic payloads of the ADC. Design details of the studies are provided by the regulatory documents issued by the FDA and the ICH.
Growth of the ADC field will bring new antibodies, conjugation technologies, and payloads. While the in vitro studies to evaluate DDI potential of the payloads are well delineated by the regulatory agencies, the impact of novel linking mechanisms on safety and efficacy of the ADC remains to be investigated. In addition to oncology, ADCs are being developed for autoimmune disorders and infectious diseases. ADCs being developed for rheumatoid arthritis carry payloads such as alendronate (anti-IL-6 ADC), siRNA (anti-C5aR1 ADC), Pseudomonas exotoxin A (anti-FRβ ADC) and glucocorticoid (anti-TNFα, ABBV-3373). Dexamethasone is a payload for two ADCs for inflammatory disorders (Vaccines 2021, 9, 1111). Some of these novel payloads, e.g., siRNA or bacterial exotoxin will not attract scrutiny regarding their effects of drug metabolizing enzymes and drug transporters. A follow-up therapeutic modality of peptide drug conjugates, represented by two FDA-approved cancer chemotherapeutics, will benefit from the experience gained in evaluating safety and DDI potential of the ADC (Molecules 2022, 27, 7232).
Conclusions
Vigilance and application of the best practices for safety evaluation of the ADC will continue to deliver life-saving therapies to critically ill cancer patients. Current DDI evaluations of the ADC are based on in vitro studies of the payload molecules. These efforts are successful, as DDI interactions in which ADC would play a role of a victim or a perpetrator did not cause withdrawal of the drugs from the market. Future ADC safety guidance developed by the regulatory agencies may contain recommendations for novel studies that are uniquely suited for this modality.






