
The Chiral Blind Spot: Why AI in Drug Discovery Must Learn to See in 3D
Valliappan Kannappan, PhD, Founder, Chiralpedia
Chandramouli Ramnarayanan, PhD, Global Technical Enablement Engineer, JMP Statistical Discovery
AI is rapidly changing Pharma and drug discovery, yet a quiet paradox remains. A molecule’s 3D handedness shapes how it heals or harms, but many advanced models are still effectively stereo-blind and can’t tell mirror images apart. This “Chiral Paradox” highlights a simple truth: unless AI gains stereochemical awareness, faster discovery may come at the expense of safety and clinical effectiveness.
Introduction:
Artificial intelligence in life sciences is attracting unprecedented investment. Generative models and deep learning platforms are positioned as tools to compress discovery timelines and expand chemical search space. Yet a core scientific constraint remains under-addressed. Many widely used AI workflows do not represent molecular chirality with sufficient fidelity.
Chirality controls biological outcomes. Two enantiomers can share the same 2D connectivity but behave differently in binding, metabolism, efficacy, and toxicity. Despite this, many current AI systems remain functionally “stereo-blind.” This creates a practical risk. If an algorithm cannot reliably distinguish a molecule from its mirror image, predictions can be misleading and, in some cases, safety-relevant.
Chirality is a known determinant of benefit and harm
Chirality has shaped modern pharmacology for decades. The thalidomide case remains the clearest reminder that enantiomers can have sharply different biological effects. Since then, stereochemical control has become a standard expectation in medicinal chemistry and regulatory science.
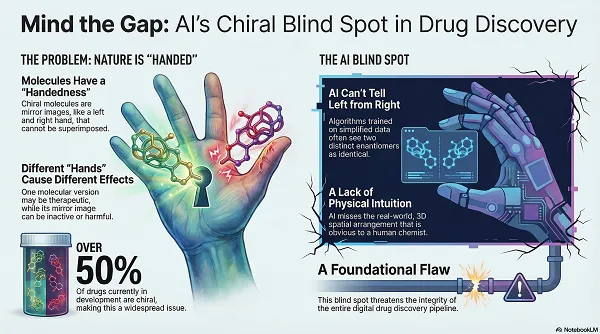
Chiral drugs are common in development and on the market. Biological systems are also chiral by construction. Proteins, enzymes, and nucleic acids form asymmetric binding environments. As a result, enantiomers often show different pharmacodynamics and pharmacokinetics, including different receptor fit, clearance pathways, and metabolite profiles.
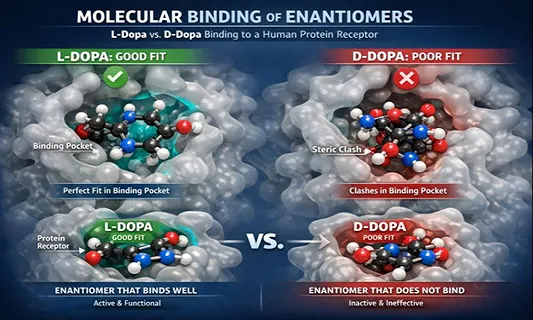
Why do many AI models miss “handedness”
The problem is not that AI cannot learn chirality. The problem is that many pipelines do not present chirality in a form the model can consistently use.
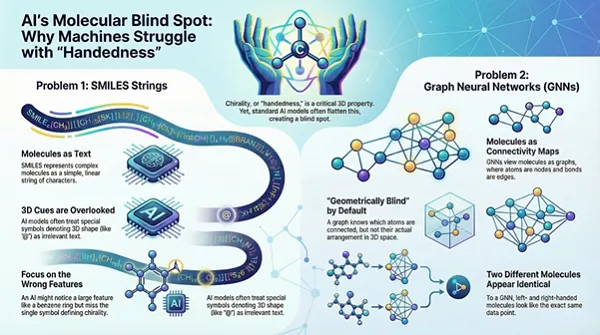
SMILES-based learning
SMILES encodes molecules as text strings. Stereochemistry can be included using isomeric SMILES conventions, but these markers can be underweighted during tokenisation and training. Models may learn strong associations with functional groups and substructures while treating stereochemical symbols as low-signal characters. That is a failure mode when enantiomer-specific outcomes matter.
Graph Neural Networks without explicit geometry
Standard GNNs represent molecules as graphs of atoms and bonds. Graph topology is not geometry. Two enantiomers typically share the same connectivity graph. Without 3D coordinates or stereochemical descriptors that the architecture can exploit, the model receives near-identical inputs for distinct physical entities.
Pipeline impact: where stereo-blindness shows up as cost and delay
Chirality errors tend to surface late, after time and budget have already been committed. The result is avoidable attrition across discovery stages.
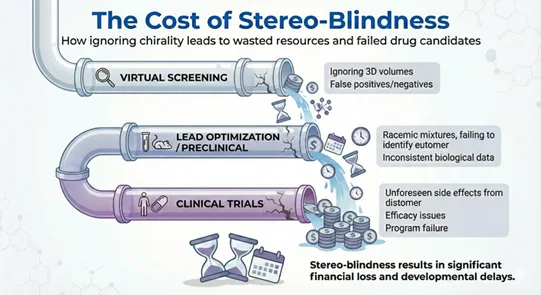
Virtual screening and docking
Stereo-blind screening can prioritise compounds that look promising in 2D or topology-based representations. In the lab, the tested enantiomer may not fit the chiral binding site. This produces false positives that consume synthesis capacity and assay time.
ADMET prediction failures
ADMET is highly sensitive to stereochemistry. Enantiomers can differ in enzyme specificity, transport, clearance, and off-target interactions. If training labels do not distinguish enantiomers, models often learn an averaged signal that does not hold for either form. That is especially problematic for toxicity risk estimation and exposure-driven safety margins.
Generative model failure modes
Generative AI can propose structures that look plausible in 2D but break under 3D scrutiny. Without stereochemical and geometric constraints, models may output candidates with infeasible stereocenters, strained conformations, or chemically unrealistic 3D arrangements. These outputs waste downstream effort and can bias decision-making toward artifacts.
Engineering “chiral intelligence”
Closing this gap requires a shift from predominantly 2D informatics to geometry-aware learning. Three approaches are emerging as practical interventions.
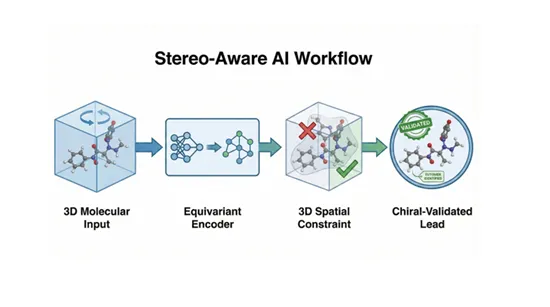
- 3D-aware architectures
Equivariant neural networks are designed to handle 3D symmetries correctly. Rotating a molecule should not change predicted properties. Mirroring it often should. SE(3)-equivariant models aim to encode geometry in a way that aligns with physical reality and molecular interaction principles. - Stereo-aware data augmentation and labeling discipline
Data quality is a limiting factor. A stereo-aware strategy includes explicit enantiomer representation and, when available, enantiomer-specific activity and ADMET labels. This forces models to learn that R and S forms are not interchangeable. It also reduces leakage where one enantiomer becomes an unlabeled proxy for the other. - Constraints for generative systems
Generative models benefit from physics-informed and geometry-aware filters. These can enforce basic stereochemical validity, feasible bond geometry, and reasonable conformational energetics before candidates are accepted for synthesis planning or ranking.
Strategic implications for R & D leadership
Chiral fidelity is not a niche technical preference. It influences late-stage failure risk, safety exposure, and portfolio economics. Industry-supported analyses have placed the cost of developing an approved drug at roughly $2.6B, with a substantial fraction tied to late attrition. Improving the fidelity of molecular recognition and ADMET prediction is one lever to reduce avoidable downstream loss.
There is also an intellectual property dimension. Single-enantiomer development of previously racemic drugs remains a known lifecycle strategy, as seen with esomeprazole following omeprazole. Organisations that can model enantiomer-specific performance earlier gain an advantage in prioritisation, differentiation, and timing.
Toward a higher-fidelity standard in digital discovery
The next phase of pharmaceutical AI should emphasise fidelity alongside speed. Autonomous molecular design will only be as reliable as the representations it uses. If molecules are treated as text strings or flat graphs, stereo-sensitive biology will continue to expose model blind spots. A more robust standard treats molecules as 3D objects operating in a chiral environment, with stereochemically explicit predictions and, ultimately, stereochemically explicit synthesis planning.
The industry message is straightforward. If AI is expected to propose drug candidates that work in real biology, it must be trained to distinguish mirror images as different molecules, because biology does.
References
- Enrico D, et al. Enantioselectivity in Drug Pharmacokinetics and Toxicity. Int J Mol Sci. 2021;22(11):5713.
- Mphahlele MJ, et al. Chirality status of medicines registered in South Africa. Sci Rep. 2020; 10:17644.
- Dufour R, et al. The significance of chirality in contemporary drug discovery, a mini-review. Drug Discov Today. 2024.
- Agranat I, et al. Chirality of New Drug Approvals (2013–2022). J Med Chem. 2024;67(4):2762-2774.
- FDA. Development of New Stereoisomeric Drugs. Guidance Document. 1992.
- Chiralpedia. Part 9: Stereochemistry in Drug Discovery and Development. 2020.
- Moores A, Zuin Zeidler VG. Don’t let generative AI shape how we see chemistry. Nat Rev Chem. 2025;9(10):649-650.
- Buntz B. How stereo-correct data can de-risk AI-driven drug discovery. Drug Discovery Trends. 15 Oct 2025.
- Chiralpedia. Episode 2: Why Chirality Confuses Machines? 2025.
- Yoshikai Y, et al. Difficulty in chirality recognition for Transformer architectures learning chemical structures from string representations. Nat Commun. 2024;15:1197.
- Wigh DS, et al. A review of molecular representation in the age of machine learning. WIREs Comput Mol Sci. 2022;12(5).
- Wang R, et al. Graph neural networks driven acceleration in drug discovery. Comput Struct Biotechnol J. 2025;23:113456.
- Li Y, et al. Efficient equivariant model for machine learning interatomic potentials. npj Comput Mater. 2025;11:45.
- Tufts Center Study of Drug Development. Cost to Develop New Drug Is $2.6 Billion. Appl Clin Trials. 2020.
- van Tilborg D, et al. Exposing the Limitations of Molecular Machine Learning with Activity Cliffs. J Chem Inf Model. 2022;62(23):5938-5951.
Disclaimer and Disclosure
The views expressed herein are personal and reflect current thinking in this domain, based on information available in the public domain. They do not necessarily represent the views of any employer, organization, or affiliated institution.
The graphics used in this material were generated using AI based on human-authored prompts and were reviewed before inclusion. However, structural, conformational, and stereochemical depictions may be incomplete, simplified, or inaccurate. Figures should be treated as illustrative concepts only and should not be used as the basis for experimental design, safety decisions, regulatory submissions, or other high-stakes scientific conclusions without independent verification using appropriate cheminformatics tools and expert review.

Valliappan Kannappan, PhD, is the Founder of Chiralpedia and a former Professor of Quality Assurance at Annamalai University, India. With over three decades of experience in medicinal chemistry, his work has focused on chiral analysis and separation techniques to address real-world challenges in pharmaceuticals and biomedical research. In 2021, he launched Chiralpedia.com to promote accessible education and collaborative research in chiral science.

Chandramouli Ramnarayanan PhD, is a Global Technical Enablement Engineer at JMP Statistical Discovery LLC, a subsidiary of SAS Institute Inc. With over 20 years of experience in academia, pharmaceuticals





