

The batch reactor has long been the mainstay of API manufacture for good reason. They are well understood, and many processes work very successfully in batch mode. Yet there are some reactions where running a batch process at a very large scale is less than ideal. High up the ‘best avoided’ list are reactions that are extremely exothermic, where running them on a large scale risks explosion in the event of a thermal runaway. But, equally, one of the reaction intermediate might be explosive, demonstrate auto-catalytic behaviour, or perhaps extremely toxic or otherwise hazardous, and handling large volumes might be considered unwise.
In such cases, the answer might lie in flow chemistry. The concept is not new, and has been touted as the potential future for the manufacture of APIs for at least a couple of decades now without making widespread inroads in replacing batch vessels. But, given the right choice of project, flow chemistry has huge potential, and has already been used at commercial scale in the manufacture of APIs and intermediates.
The key difference between a continuous flow reactor and a batch reactor is, of course, the volume of reagents that reside within the vessel at any one time. Batch reactors with a 10,000L capacity are not uncommon in a commercial environment, whereas the volume within a flow reactor might be as small as a few millilitres. In a batch process, the reaction mixture will usually remain within the reactor for several hours, whereas in a flow reactor, it will pass through quickly – the residence time is commonly a handful of seconds or a few minutes – and heat exchange is drastically increased. This short residence time also means the product is far less likely to decompose, or to over-react and form side products.
Work-up can also be carried out in flow if another device is connected in series, with the reaction mixture flowing from the reactor device directly into a work-up device to quench the reaction. Multiple reaction steps can even be done in sequence, by dint of connecting another reactor (or reactors) after the first one.
Scaling up can be achieved simply by leaving a continuous flow reactor running for longer, and, if necessary, running multiple reactors in parallel. This represents a further advantage: when scaling up a batch process, it is normal to sequentially scale up through multiple, increasingly large, vessels, with tweaks to the reaction conditions commonly being necessary. It is, of course, also possible to create a production-scale reactor that is a little larger than the one used in development to reduce the need for parallelisation.
With a flow process, the reaction conditions for an experimental scale will likely be identical to those for pilot scale and full-scale commercial production. This will speed up the overall development process, as much of the time required for optimisation is eliminated.
Reaction types
As a rule-of-thumb when trying to decide what type of reactor to use for a particular process when switching from batch to continuous, the nature of the reaction should be considered carefully. Reaction characteristics can be split into three types, depending on their reaction rates and behaviour under standard operating conditions. Type A reactions proceed extremely quickly – they will have a half-life shorter than one second, and the rate will be controlled by mixing or mass transfer.

A type C reaction is much slower, typically taking 10 minutes or more to go to completion. The timescale can even be an hour or more. These slow, kinetically controlled reactions might benefit from being carried out in flow if there were safety considerations in play, or if they are amenable to process intensification at higher temperature, pressure, reagent concentration, or a combination of these factors.
Between the two lies Type B, with the reaction proceeding in seconds or minutes. These reactions are, predominantly, kinetically controlled, but it may be that they are limited by mass transfer. Efficient heat removal and good mixing will both be necessary but in many cases a good control of residence time may also be required at time scale that is not achievable in a batch reactor. (It is often possible to speed them up by increasing pressure, temperature, reagent concentration, or a combination of these factors).
Reactor types
There are several basic types of flow reactors. Coil reactors – essentially a long tube, usually coiled up to resemble a spring – are most suitable for homogeneous processes. These are most likely to be appropriate for Type C reactions, and some Type B reactions, and can improve safety via process intensification.
Type A reactions are more likely to be successful if they are run in some form of plate reactor, where the reaction takes place in a channel cut into the plate. This may be a good option if mixing control is beneficial, and the reaction time ranges from milliseconds to seconds. It should be possible to increase the yield with better mixing or improved heat exchange, or both, and the correct design of reactor can prevent the reaction from overcooking. Some Type B reactions are also best carried out in a plate reactor.
If the reaction is heterogeneous, then a packed bed reactor may be appropriate; metal catalysts, for example, can be coated onto a support within the reactor. These vessels are usually made from stainless steel, or even Hastelloy, enabling reactions to be carried out at higher pressures. And then there is the continuous stirred tank reactor, which uses active mixing such as an impeller, which can also be useful for heterogeneous processes.
The physical form of the reagents has an impact on reactor design, too. Liquid–liquid reactions benefit from some form of mixing strategy within the reactor; gas–liquid reactions will need some form of pressurised reactor. But in both cases, either a plate or a coil design could be used, with the precise selection depending on the reaction type.
Reaction condition choices
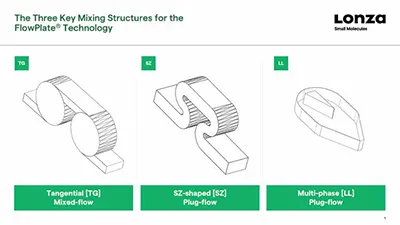
Mixing is an important consideration when designing a flow process. If active mixing is not used, then some form of passive mixing will be required to ensure the different components of the reaction come into proper contact with each other. This could be as simple as two streams of reactants entering the reactor at steady flow rates via a T-piece, but if more effort is required to make them mix effectively, then structures within the reactor that cause turbulence may be required. Options include a tangential design, SZ-shaped curves, or a more complex liquid-liquid design for multi-phase applications.
Temperature and pressure also need to be considered. Temperature is far easier to control within a flow reactor than it is in a large batch vessel, with the larger surface area giving better heat transfer. This is particularly important when thermal runaway is a possibility, one of the reasons why some reactions are particularly hazardous in batch.
Pressure can be important, too. If one of the reagents is a gas, for example, then it can increase its solubility, or its speed of dissolution, in the liquid reaction mixture. Pressure can be increased via a back pressure regulator.
With a pump of some form used to introduce the reagents into the reactor on a continuous basis, the flow rate is an important factor in controlling the reaction. This, and the length of the path through the reactor, will affect the residence time – the amount of time the reaction mixture spends within the reactor before it emerges from the other end. The time needs to be sufficiently long that the reaction goes to completion, but not so long that side-products start to form or the product begins to decompose in some way, for example.
Hazardous reactions
Of course, before any process is moved from batch to flow, a comprehensive assessment of the reagents, reactor type and reaction conditions will have to be carried out to determine the optimal design. A great example of a study on continuous flow in hazardous chemistry comes in the form of a recent collaboration between Lonza experts and scientists from the Center for Continuous Synthesis and Processing and the University of Graz in Austria. The aim was to see how effectively sulfonyl chlorides could be made using N-chloroamides, and a variety of reagents and reaction conditions were investigated.
Organic sulfonyl chlorides are the starting point for a variety of different sulfonyl derivatives including sulfonamides, which are of particular importance in the pharmaceutical industry. These groups are common in drug structures, featuring in medicines as diverse as antibacterials, HCV polymerase inhibitors, beta-3 receptor agonists, and even carbonic anhydrase inhibitors.
Typically, sulfonyl chlorides are made by the oxidative chlorination of either a thiol or a disulfide, a process that normally uses chlorine gas in the presence of an acid. Using chlorine gas rather than a chlorinating reagent is extremely atom-efficient, but its hazardous and gaseous nature means great care is required in handling it, so alternative chlorinating agents are commonly used, but many of these have limited functional group tolerance, and they are often poorly selective.
N-Chloroamides represent another alternative. These reagents, such as N-chlorosuccinimide (NCS), 1,3-dichloro-5,5-dimethylhydantion (DCH) and trichloroisocyanuric acid (TCCA), are milder, and tend to make fewer side products. But they have a downside, particularly at scale: they are thermally unstable, and if reactions are carried out at elevated temperature, they can be explosive. They therefore represent an ideal candidate for continuous flow chemistry, with microreactors ensuring that only small volumes of reaction mixture are present at any one time, alongside a high surface to volume ratio. Could we create a flow chemistry protocol to make sulfonyl chlorides using N-chloroamides?
The simple answer is: yes we could. We selected DCH as this gave a homogeneous solution throughout, preferable for flow reactions, and found that in the reaction with diphenyl disulfide, 2.5eq of the reagent were required to make the reaction go to completion. Our initial studies on the reaction suggested that it could be considered either Type A or Type B, depending on the reaction conditions selected. We therefore chose a microstructured plate flow reactor that was designed to give efficient heat exchange and good plug-flow mixing, via an SZ mixing structure.
We found that increasing the residence time from 24s to 48s upped the yield from 45% to 87%. We also realised that results from the SZ mixing plate showed the reaction could actually be categorised as Type B, so in order to overcome the corrosion issue observed with metallic plates (even Hastelloy C showed corrosion at higher temperature), a simple T-piece followed by a coil out of polymer material ought to suffice. Using this system, and increasing the residence time further to 58s, gave close to a quantitative yield. Similar protocols also worked successfully with a variety of other disulfide substrates, and also with thiols
Temporal kinetic profiling was then used to pinpoint the auto-catalytic nature of the reaction and appropriate operating conditions for a preparative scale process (the batch results had shown a reaction runaway even at laboratory scale). These were checked out with a four-hour run, and a throughput of 4.3g/h was achieved, with benzendulfonyl chloride isolated in 81% yield, and with a purity above 98%. We even managed to telescope the reaction into an amination reaction, by adding a T-piece to introduce diethylamine in acetonitrile, plus a second coil where the amination reaction took place. This successfully formed a sulfonamide, in 94% isolated yield with a 156s residence time.
Towards a larger scale
These experiments clearly show the potential for the synthesis of sulfonamides via continuous flow to be scaled up and produce large quantities of material. By tweaking multiple parameters, including the reactor style and set-up, reagent stoichiometry, temperature and residence time, very high yields of pure products can be achieved for reactions that present significant hazards when run in batch, including the avoidance of thermal runaway.
As an example, we scaled up the continuous flow manufacture of an indole compound. This was required on a large scale as a starting material for the synthesis of an API. The reaction was run at very high temperature and pressure – 285°C and 80 bar – with a coil-shaped reactor enclosed within a thermal fluid calendar. Two of these were connected in series, and we managed to produce 2,125kg of the compound, with an 81% yield, in 32 days. The material was in spec, and was used successfully in the next step of the reaction sequence to make the API.
In a many cases batch processing remains the method of choice for many reactions where the chemistry is straightforward and the reagents, solvents and products do not present particular hazards. But for the right process, flow chemistry represents a simple way to make and scale up a reaction, especially those that are, one way or another, hazardous, but also increasingly more standard chemistries where sufficient process intensification can be achieved.
References:
1. P. Plouffe, A. Macchi and D.M. Roberge, Org. Process. Res. Dev. 2014, 18, 1286
2. D. Polterauer et al., React. Chem. Eng. 2022, 7, 2582





