

Mass Spectrometry (MS) is playing a bigger role than ever in supporting biopharmaceutical CMC development. This is in large part due to advances in MS instrumentation, more separation techniques becoming amenable to MS detection and advancements in data processing and system operation software. MS really has had a face lift. It was once thought of as niche technique, needing highly skilled operators to both operate the systems and also process the data, and only used for the extended characterisation of biopharmaceuticals and bioanalysis. Such limitations led to minimal use of MS in biopharmaceutical development and data was rarely included in regulatory dossiers. One of the biggest barriers to using MS data in a QC release environment is the requirement for FDA 21 CFR Part 11 compliance. FDA 21 CFR Part 11 governs the use of electronic records and signatures; MS techniques generate large data sets containing significant amounts of meta-data which all needs to be tracked. In recent years, advances in this area have allowed the use of MS data in QC. Now multiple MS vendors offer 21 CFR Part 11 compliant solutions for both operating systems and data processing options.
New MS hardware options have led to an ever growing list of workflows that can be employed to support biopharma development. The introduction of softer ionisation techniques and native MS have expanded it usage in workflows such IEX (Ion exchange chromatography)-MS and SEC (Size exclusion chromatography)-MS. The key improvement being the speed at which the scientist gets the data required to make critical product development decisions. Workflows that used to require weeks of data analysis can be now completed in minutes. Currently multiple vendors offer MS vendor neutral software enabling the harmonisation of workflows and offering further reductions in data processing timelines. These software solutions allow data from multiple workflows to be brought together enabling a holistic view of the molecule.
MS has increased in popularity due to its flexibility to work with multiple different product types from antibodies through to cell and gene therapies. The ability to setup platform workflows across modalities is advantageous compared to other analytical methods. The simplicity of both hardware and software allows workflows to become routine in non-specialist MS labs, i.e process development for PAT, process analytical and QC laboratories.
In this article we focus on two of the most significant areas of development in recent years for MS workflows; Multi Attribute Methods (MAM) and Host Cell Protein (HCP) analysis.
Multi-attribute method –
The term Multi-attribute method was first coined by Amgen and is now widely adopted in the biopharmaceutical industry. As originally proposed the MAM approach is in essence a peptide mapping analysis by LC MS/MS but the novelty is in its application to monitor multiple product attributes in a single analysis.
The idea of using a single method to monitor a range of attributes was born from the shift to (Quality by Design) QbD approach recommended by regulatory agencies. The QbD approach includes establishing a quality target product profile (QTPP) that identifies potential critical quality attributes (pCQAs). Once determined the pCQAs need to be consistently monitored throughout development and production. For this reason, advanced analytical approaches are needed to ensure a CQA driven manufacturing process instead of final product testing. The MAM approach supported by recent advancement in MS has the potential to meet this need.
Execution of MAM usually begins by digesting the protein of interest with site-specific protease (trypsin is most commonly used) followed by chromatography separation of the resulting peptides and their detection by MS. The peptides undergo two stages of MS analysis: MS1, where their accurate mass is determined and MS2, where the peptides are fragmented to reveal their amino acid sequence. The MS data is analysed by specialised software to produce the primary amino acid sequence of the protein as well as any modifications (e.g. deamidation, oxidation, glycosylation). The MS1 data can be used to quantify the relative abundance of the detected modifications while the MS2 can provide site-specific localisation of the modification. Another advantage of MAM is that it can be used to detect unmonitored quality attributes (New Peaks) and thus identify potential process and product-related impurities. As post-translational modifications and impurities could have effect on product safety, activity or immunogenicity they are potential CQAs and their detection and identification is essential throughout the product life cycle.
Advantages of MAM
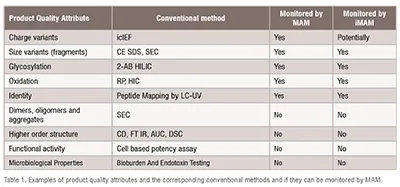
Traditionally, a series of methods are used to characterise the product CQAs e.g. icIEF for changes in charge variants, 2-AB labelling and HILIC separation for glycans or Reverse Phase for oxidation etc. (see Table 1).
If we consider the time required to develop, validate and transfer these methods as well as execute them this approach is not supportive of accelerate product development. Moreover, the conventional analytical methods address categories of product related variants e.g. charge variants but cannot distinguish e.g. between different deamidation sites and their individual contribution to the observed variant. MAM provides site-specific information often down to specific amino acid thus allowing for enhanced product understanding.
MAM also outperforms other methods when it comes to detection of New Peaks. New Peaks can sometimes be observed by current purity tests, but as these are limited to UV based detection they can easily be misled by co-eluting components. MAM is more sensitive than UV based approaches and is able to further investigate and assign identity to the unknown components by applying the principles of “Error Tolerant Searching” and de novo sequencing.
Overall, these unique capabilities make MAM an enabler of the QbD approach

iMAM
Most publications refer to MAM as peptide level analysis following protease digestion. However, MAM can also be performed at the intact protein level (iMAM). In the recent years technological advances in the field of native MS have allowed this approach to become a reality. MS of intact proteins under native conditions used to be state-of-the-art approach employed only by a few specialised laboratories around the world. Since instruments with extended mass range became commercially available this approach has become more accessible to any laboratory with expertise in MS. As a result native MS is now beginning to be applied for the characterisation of biotherapeutics. The approach has the advantage of limited sample preparation where majority of the variability of MAM stems from. iMAM offers great potential for characterising the heterogeneity of glycosylated biotherapeutics and for supporting higher order structure and non-covalent interaction studies. Despite that this is a relatively new approach with only a few documented examples in the literature it is already being applied for product quality testing and to couple common separation methods such as SEC and IEX to MS.
MAM as a method for QC
MAM coupled to LC MS/MS has been proposed as a replacement for several physicochemical QC methods used for batch release or stability testing. However, the intention is not to replace all QC assays with MAM. As shown in Table 1 detection of dimers and aggregates, higher order structure, biological activity and microbiological properties are outside the remit of MAM and will continue to be monitored using conventional methods. Despite that, there aren’t any regulatory impediments to introduce MAM in QC recent publications from FDA emphasize that majority of BLA application include MS data but none as part of release or stability testing. The main bottlenecks appear to be the need for bridging studies, the lack of MS expertise in QC labs and complexity of the validation. Perhaps the MAM approach will be most successful in the instances where no conventional methods have been applied to control the molecule. (Table 1)
Host cell protein (HCP) analysis
Host cell protein content is critical quality attribute that can impact product safety and efficacy and monitoring clearance is obligatory. Traditionally ELISAs have been the method of choice for measurement of HCP. But there has been a paradigm shift and last year the first ever HCP-MS method was approved for product release testing to support a phase III clinical trial (1st MS-based HCP analysis under GMP for release test (alphalyse.com)). The COVID pandemic accelerated CMC timelines for new medicine approval became the norm and this is a challenge when it comes to developing a product and process specific ELISA. Ease of development makes the use of a MS based method advantageous for supporting shorter CMC timelines. The appropriate method for host cell protein analysis needs to be considered as part of the overall process and analytical risk assessment, and there is now flexibility, due to improvements in MS hardware and software, to employ MS as the sole analytical method for control of HCP. This is no better illustrated in an update to the United States Pharmacoepia (USP) chapter 1132, there is a new chapter USP 1132.1; Residual Host Cell Protein Measurement in Biopharmaceuticals by Mass Spectrometry, that contains general guidance and best practices for MS-based identification and quantification of high-risk and high-abundance HCPs.
Enabling MS technolgies to support host cell protein analysis by MS
The MS requirements from a hardware and software perspective for HCP analysis are a different prospect compared to a MAM workflow. Whilst a high resolution and high sensitivity sensitive MS system is a definite benefit to a MAM workflow, it is essential for an MS HCP screening method. The advances in MS sensitivity have brought the levels of sensitivity to comparable levels of a HCP ELISA, in the order of magnitude of part per million (ppm). Such sensitivity was needed to enable to use of MS. It is also important to note that with any MS assay, as with any ELISA, the sensitivity is HCP dependent. For any HCP MS workflow the MS sensitivity and resolution is only one factor, it is a careful balance between separation technique and amount of sample loaded. Chromatographic separation is most commonly used prior to MS HCP analysis but it is possible to use CE based separation methods. Advances in chromatography systems and columns have also significantly improved the repeatability and robustness of the HCP MS workflow. Depending on the HCP MS workflow different softwares are required, but now there are multiple options for software depending on the workflow. The key is matching the HCP MS workflow and software with what is needed from an analysis perspective.
What method to choose for HCP-MS
Advances in MS hardware and software in the last ten years have provided flexibility in terms of biopharma HCP MS workflows. Normally there will be a discovery or screening workflow to identify as many host cell proteins with relative quantitation, followed by a more targeted method that yields better quantitation and sensitivity. There is a trade of between a method with maximum sensitivity and one with the highest level of accuracy for quantitation, the decision on the workflow should always be based upon the question that needs to answered.
Conclusion
In summary the number of MS workflows in routine and ad hoc use for biopharmaceutical CMC development has never been higher and will only continue to grow given the increasing advances in MS instrumentation and associated separation techniques. Here we have illustrated through two of the most popular MS workflows MAM/iMAM and HCP-MS how far the use of MS has come in the last 10 to 15 years. The major challenge is still data processing given the large amount of high quality data produced by modern MS intruments, and how to interpret this, we are only really scratching the surface of utilising all the data is available. MS software vendors need to embrace AI and machine learning tools in order to help develop software that process MS data quicker, more easily and providing even more valuable insights to help accelerate biopharmaceutical CMC development.







