


Driving Biotech Innovation
Navigating Fast-to-Clinic and Fast-to-Market Strategies from Discovery to Commercialization
Kishore Kumar Hotha, Global Vice President, Veranova
This article explores biotech's fast-to-clinic/market strategy, focusing on the role of CDMOs. It emphasizes the strategic decision-making and phase-appropriate approach that streamlines R&D, the importance of flexibility and agility in adapting to regulatory and market changes, and the significant role of CDMOs in enhancing product development speed without sacrificing quality.

In the dynamic landscape of pharmaceutical research, small- to medium-sized biotech firms, many of which operate virtually, play a crucial and integral role in early drug development. These firms focus on reaching key milestones, such as submitting investigational new drug (IND) applications or leveraging intellectual property for acquisition deals. Given their unique goals, constraints, and regulatory requirements, their approach to Chemistry, Manufacturing, and Controls (CMC) must be highly customized. The strategic use of external advisors and contract manufacturing organizations (CMOs) is critical in crafting CMC strategies tailored to each company's needs.
Transitioning from discovery to market is a complex journey, with only about 6% of molecules progressing to Phase III clinical trials. This high early attrition rate, often due to CMC complexities, underscores the need for strategic planning to navigate the CMC landscape from development to commercialization effectively. A well-thought-out strategy can provide reassurance and confidence in facing these challenges. The rapid development of COVID-19 vaccines, achieved in less than a year, serves as a beacon of hope, demonstrating the potential to expedite development phases while upholding safety and compliance standards.
The 'Fastlane' strategy, designed to reduce market entry times, involves proactive planning and resource allocation to promising drug candidates. However, the need for speed must be carefully balanced with the responsibility to ensure patient safety and regulatory compliance. Diligent attention to these aspects is crucial to minimize financial risks and enhance the likelihood of clinical success (Figure 1).
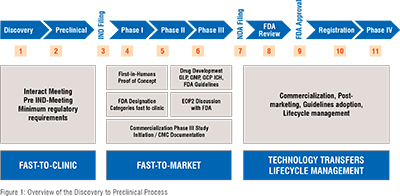
NAVIGATING FDA GUIDELINES FOR RAPID CLINICAL ENTRY:
The FDA's five special designations—Orphan, Fast Track, Accelerated Approval, Priority Review, and Breakthrough Therapy—accelerate the development and approval of medications for severe medical conditions with unmet needs, benefiting healthcare providers and patients. Orphan Designation targets drugs for rare diseases affecting fewer than 200,000 people in the U.S., offering benefits like tax credits and extended exclusivity. Fast Track facilitates faster development for severe conditions lacking therapies by enabling more frequent FDA interactions and a rolling review process. Accelerated Approval allows drugs that improve on existing treatments through surrogate endpoints to enter the market sooner, pending confirmation in post-marketing studies. Priority Review shortens FDA review times from ten months to six for drugs with significant treatment advances, while Breakthrough Therapy offers all Fast Track benefits plus intensive FDA guidance for drugs that markedly improve on existing therapies, ensuring rapid and efficient market entry.
OPTIMIZING IND SUBMISSIONS FOR FAST-TRACK APPROVAL
The requirements for an Investigational New Drug (IND) submission are critical in the fast-to-clinic approach, demanding a comprehensive dossier that aligns with the strict regulatory standards of the U.S. Food and Drug Administration (FDA). This dossier includes detailed preclinical study data such as pharmacological, toxicological, and pharmacokinetic evaluations to affirm the investigational product’s safety and justify human trials [8,9]. It also incorporates Chemistry, Manufacturing, and Controls (CMC) information detailing the synthesis, characterization, and quality control of the drug substance and product, ensuring their consistency and safety. Moreover, the submission must present a proposed clinical trial protocol that outlines the study design, objectives, and methodology, along with credentials of clinical investigators and informed consent forms for participants, making it a pivotal step in advancing to clinical trials.
Accelerating the drug development process involves optimizing supply chains, refining project management, implementing automation, outsourcing, and efficiently managing inventory. Emphasizing early coordination between biotech companies and Contract Development and Manufacturing Organizations (CDMOs) is vital. This strategy leverages existing data, enhances process optimization, and promotes early engagement with regulatory bodies. Early attention to CMC activities, streamlined synthesis, and formulation development are essential to tackle manufacturing challenges. Adaptive trial designs and strategic regulatory pathways like Fast Track or Priority Review can substantially shorten development timelines. Robust project management and outsourcing to CDMOs expedite progress by drawing on external expertise, ensuring that development plans are robust, scalable, and comply with regulations, thus accelerating the journey from discovery to clinical trials.
Accelerating from IND to NDA: Clinical Study Strategies
Following IND approval, phase 1 clinical studies assess human safety and efficacy, involving healthy volunteers and requiring a strategy finely tuned to drug development specifics. This initial phase focuses on process optimization and control within the CMC sections, including revisiting existing data, assessing manufacturing capabilities, leveraging technical expertise, optimizing processes, and implementing impurity controls. These activities must be meticulously calibrated, considering clinical significance, drug designation, and the urgency of a fast-to-clinic approach, with essential coordination with CDMOs.
Safety and efficacy are paramount at all stages, necessitating clear expectations and prioritizing streamlined activities to align with regulatory standards and ensure the delivery of safe, effective therapeutics. A focused review of CMC development across phases is vital from IND to NDA. Early phase requirements establish a foundation for safe drug development, while later phases demand rigorous validation and scalability to meet regulatory standards for widespread use.
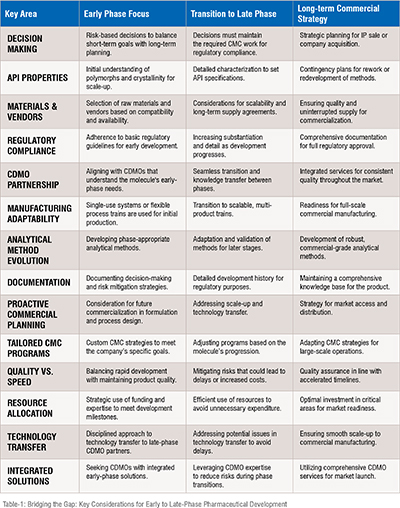
MASTERING CMC CHALLENGES IN EMERGING BIOTECH VENTURES
For emerging biotech companies, mastering the complex landscape of Chemistry, Manufacturing, and Controls (CMC) development is pivotal in transitioning from early-stage innovation to commercial success. Early phases focus on establishing a solid foundation through meticulous risk mitigation and adherence to regulatory and industry standards without compromising essential CMC activities. These stages are crucial for accurate toxicology assessments and a thorough understanding of the Active Pharmaceutical Ingredient (API) solid-state properties. Scalability in formulating processes is essential to ensure seamless transitions from clinical to commercial stages.
As development progresses to late-stage and commercial phases, the need for scalability and advanced analytical methods becomes more pronounced. Transitioning from disposable to multi-product, commercial-scale process trains accommodate growing production demands. This stage requires evolving analytical techniques to be robust and stable, indicating protocols crucial for maintaining regulatory compliance and ensuring product stability. Comprehensive documentation throughout this phase facilitates smoother regulatory reviews and approvals, streamlining the transition into commercial manufacturing.
A strategic partnership with a Contract Development and Manufacturing Organization (CDMO) is a cornerstone of the CMC development journey. Selecting a CDMO that aligns well with the molecule's requirements from early to late phases can significantly enhance operational speed, reduce overhead, and ensure streamlined knowledge transfer. These collaborations are tailored to address the unique challenges of each phase, recognizing that a one-size-fits-all approach is insufficient. By planning early, meticulously documenting development processes, and customizing CMC activities to fit specific timelines, funding limitations, and regulatory frameworks, biotech companies can navigate the path to commercialization with greater assurance and efficiency.
Strategic financing in biotech development critically shapes the operational decisions of small biotech companies. These organizations must navigate the delicate balance between essential development activities that cannot be postponed and those that can be temporarily deferred when funding is limited. This balancing act is crucial as securing adequate financing supports ongoing development and ensures that regulatory activities, which are vital for long-term success, are not compromised. Practical strategies might include prioritizing core development activities directly impacting regulatory success and exploring alternative funding sources, such as partnerships or venture capital, to sustain other necessary operations without delay.
Navigating regulatory landscapes for drug approval involves regulators requiring comprehensive data detailing the synthesis or fermentation of the drug substance, along with its characterization, testing methodologies, and stability. Drug product formulations and phase-appropriate stability tests must also be established and documented. In practice, biotech companies must invest in thorough early-phase testing and data collection to build a robust foundation for regulatory submissions, ensuring that each step of the process is well-documented and meets the stringent requirements of regulatory bodies.
Optimizing CMO partnerships is crucial for small biotech firms that rely on these organizations to handle their CMC activities. This outsourcing demands meticulous management to keep the development timeline on track. This involves establishing clear communication channels and setting precise expectations with CMOs to ensure all parties understand the timelines and critical milestones. Managing these relationships effectively is crucial, as delays or miscommunications can directly impact the supply of clinical trial materials and the support of subsequent applications.
Tailoring development strategies to clinical phases and balancing speed and quality are crucial in the competitive and fast-paced pharmaceutical industry. Biotech companies sometimes need to accept calculated quality risks to accelerate development phases. However, by gathering comprehensive data before critical stages such as Phase III trials, companies can improve the efficiency of these trials and potentially reduce overall development time. This involves strategic decision-making to determine when accelerated timelines are worth the risk and when more thorough data collection will benefit the project in the long run.
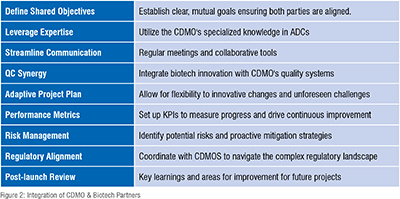
ACHIEVING CLINICAL SUCCESS: LEVERAGING CDMO CAPABILITIES
Streamlining Clinical Development: Timelines and Requirements
The development timing and requirements for progressing a pharmaceutical product from preclinical stages through commercialization are meticulously planned to ensure efficiency and compliance with quality standards. Initially, the process involves a familiarization run-through on a small scale, typically less than 10 grams, which requires about one week per step across different development stages. As the scale increases, from kilo scale preparation (up to 100 liters) in both GMP and non-GMP environments to plant manufacturing, additional time is allocated for scaling up and ensuring quality, with increased time needed for plant manufacturing processes across all stages.
In preclinical and Phase I, the approach maintains a high-risk tolerance, emphasizing identifying key quality attributes of raw materials and tracking critical impurities. The process is designed to ensure purity through chromatography and stability under standard conditions. The requirements become more stringent as the product moves to Phase II and III. The focus shifts to optimizing the process through experimental verification, vendor qualification, and detailed design of experiments (DOE). The process parameters are increasingly optimized, and major impurities are managed with strategic hold points and assessments. By the commercial phase, the process is fully optimized to ensure robustness, minimal waste, and optimal yield, with all impurities identified and controlled, providing the drug’s stability and efficacy for market release.
This structured approach ensures compliance with regulatory standards and maximizes efficiency and productivity(Figure 2).
Efficient Pathways for Rapid Market Entry
A strategy focusing on rapid progression from initial development to clinical stages emphasizes clarity and cost control. It involves specific plans for the scope of work (SoW), including timelines and pricing, to ensure projects stay within budget. By adopting flexible approaches in the discovery phase and selecting the most viable formulations early on, the process seeks to minimize later modifications, speed up the development, and ensure intellectual property can be appropriately secured.
Tailored Solutions Driving Cost Efficiency
The approach leverages different costing models that clarify deliverables, timelines, and costs, thereby maintaining budget adherence. It features flexible solid form exploration early in the discovery phase, accelerating familiarization and scale-up processes. This strategy ensures a smoother regulatory development trajectory and caters to specific program needs through customizable analytical and chemical development options, enhancing cost-effectiveness.
Strategic Approach for Early-Phase Market Penetration
The commercial strategy is designed to attract early-phase clients through a well-structured proposal review and pricing strategy. It introduces a standardized template for SoW and a pricing model to leverage the project's total cost to provide more attractive early-phase proposals. It incorporates comprehensive chemical and analytical development strategies that streamline processes, ensure quality control from the onset, and facilitate a faster transition from development to clinical stages (Figure 3).
Phase-Appropriate Solutions for Market Expansion
The holistic offering is designed to expand the customer base and build opportunities through a phase-appropriate approach. It integrates a standard proposal review process to ensure alignment with strategic objectives, optimized early developability assessment to lock in preferred forms, streamlined development to reduce process times, and tiered analytical development options that align with client needs. The production strategies are clarified early to ensure quality assurance and process control, complemented by a fixed fee pricing model that provides clear cost expectations.
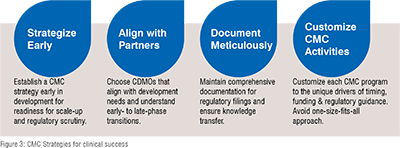
Conclusion
Designing a phase-appropriate CMC (Chemistry, Manufacturing, and Controls) strategy is crucial for the success of emerging biotech companies. These strategies must be tailored to balance the company's unique drivers and goals with the specific product requirements, as the "one-size-fits-all" model does not apply. Each CMC program must be individually crafted to navigate the complex interplay of funding, regulatory expectations, and the practicalities of drug development. This strategic approach is essential as the pharmaceutical industry evolves, playing a critical role in a company’s ability to bring new therapies to market successfully. Emerging biotech companies face unique challenges that require bespoke CMC strategies. By focusing on a phase-appropriate approach and emphasizing careful planning and partnerships, these companies can navigate drug development complexities swiftly and successfully, bringing their innovations to market.
References:
1. Navigating the Small Molecule CMC Pathway: Strategies for Avoiding Regulatory Hur-dles and Early Drug Development Pitfalls. (2023). Retrieved from https://www.xtalks.com
2. Sutherland, J. J., Woodhead, J. L., & Longo, J. (2021). Exploring different approaches to improve the success of drug discovery and development projects. Future Journal of Pharmaceutical Sciences, 7(1), 25. https://doi.org/10.1186/s43094-021-00268-4
3. Phase-Appropriate CMC Activities Facilitate the Transition from Early Development through Commercialization. (2023). Retrieved from https://www.pharmasalmanac.com
4. Moriconi, A., Stranges, D., Strodiot, L., Tello Soto, M., Zwierzyna, M., & Campa, C. (2023). CMC strategies and advanced technologies for vaccine development to boost acceleration and pandemic preparedness. Vaccines, 11(7), 1153. https://doi.org/10.3390/vaccines11071153
5. Preclinical Development of New Medicines. (2023). McKinsey & Company. Retrieved from https://www.mckinsey.com
6. CMC in Drug Development and Life Cycle Management. (2023). ICON plc. Retrieved from https://www.iconplc.com
7. Pammolli, F., Righetto, L., Abrignani, S., Pani, L., Pelicci, P. G., & Rabosio, E. (2020). The endless frontier? The recent increase in R&D productivity in pharmaceuticals. Journal of Translational Medicine, 18(1), 162. https://doi.org/10.1186/s12967-020-02313-z
8. Michaeli, D. T., Michaeli, T., Albers, S., Boch, T., & Michaeli, J. C. (2023). Special FDA designations for drug development: Orphan, fast track, accelerated approval, priority review, and breakthrough therapy. The European Journal of Health Economics. https://doi.org/10.1007/s10198-023-01639-x
9. U.S. Food and Drug Administration (FDA). (2023, September 28). Understanding investi-gational drugs for patients. Retrieved from https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers
10. The Basics of Preclinical Drug Development for Neurodegenerative Disease Indications. (2023). BMC Neurology. Retrieved from https://bmcneurol.biomedcentral.com
11. U.S. Food and Drug Administration. (2020). Investigational new drug (IND) application. Retrieved from https://www.fda.gov/drugs/types-applications/investigational-new-drug-ind-application
12. U.S. Food and Drug Administration. (2023). Overview of FDA expedited programs for se-rious conditions. Retrieved from https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/overview-fda-expedited-programs-serious-conditions
13. Marks, P., & Gottlieb, S. (2018). Balancing safety and innovation for cell-based regener-ative medicine. The New England Journal of Medicine, 378, 954-959. https://doi.org/10.1056/NEJMsb1710591
14. Ventola, C. L. (2015). The drug approval process: Accelerating approval pathways. Pharmacy and Therapeutics, 40(5), 334-338. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4418673/
15. Shetty, Y. C., & Marathe, P. A. (2019). Accelerating pharmaceutical development through the fast track, breakthrough therapy, and priority review designations. Ameri-can Journal of Therapeutics, 26(3), e344-e352. https://doi.org/10.1097/MJT.0000000000000732
16. Johnson, J., & Stevenson, R. (2021). Enhancing IND approval processes through strategic CMC practices. Journal of Pharmaceutical Innovation, 16(3), 223-237. https://doi.org/10.1007/s12247-021-09509-x
17. CMC in Drug Development and Life Cycle Management. (2023). ICON plc. Retrieved from https://www.iconplc.com
18. Davis, S., & Thompson, R. (2021). Enhancing pharmaceutical development through cus-tomized solutions. Journal of Commercial Biotechnology, 27(3), 34-45. https://doi.org/10.5912/jcb894
19. Evans, W. G., & Patel, A. (2019). Strategic approaches to early-market advantages in bi-opharma. Biotechnology Advances, 37(6), 107384. https://doi.org/10.1016/j.biotechadv.2019.107384
20. Kim, Y., & Zhao, X. (2020). Expanding market base through comprehensive biopharma strategies. Journal of Business Venturing, 35(5), 105982. https://doi.org/10.1016/j.jbusvent.2020.105982
Author Bio

Dr. Kishore Hotha is a scientific & business leader in the pharmaceutical biotech & CDMO. He contributed several IND, NDA & ANDAs of drug substances and products in small and large molecules, ADCs, Oligos, and peptides. He holds a Ph.D. from JNT University and an MBA from SNHU. Currently serving as Global VP at Veranova, his career includes pivotal roles at Lupin and Dr. Reddy’s, and he has contributed to over 50 publications and serving editorial boards.

")
